

The “Regulatory Gap”
in LED Therapy
If a consumer brand already paid for “independent testing,” why won't they apply for global medical clearance? The answer reveals an industry loophole that puts your skin at risk.
One day. One device. One test.
Confirms the battery won't catch fire. Zero ongoing accountability. No clinical proof required. No obligation to report injuries.
GAP
Continuous. Legal. Enforced.
Ongoing unannounced audits. Mandatory adverse event reporting. Peer-reviewed clinical proof of efficacy. Verified biocompatibility.
“Third-Party Independent Testing” is a marketing phrase describing a one-time electrical safety snapshot. It carries zero ongoing legal accountability. FDA Class II Medical Device clearance requires unannounced manufacturing audits, rigorous clinical substantiation, and a legal obligation to report any adverse effects — standards most consumer gadget brands cannot pass.
In the rapidly expanding Red Light Therapy market, the phrase “Third-Party Independent Testing” is heavily weaponised in consumer marketing. Brands display lab reports to build superficial trust — but this actively distracts from the most critical question in photomedicine.
The Crucial Industry Question
“If your technology is genuinely safe and effective enough to pass a third-party lab test, why haven't you submitted those exact results to the FDA for official, legal medical clearance?”
The unspoken answer is the Regulatory Gap: medical regulators demand sustained manufacturing precision, longitudinal safety data, and verified clinical evidence that most consumer “gadget” companies cannot afford or achieve.
What True Medical Regulation Actually Requires
A third-party test is a singular event. It means an independent lab looked at one golden-sample device on one day to confirm the battery wouldn't catch fire. The company has no legal obligation to maintain that standard — and no obligation to report if the LEDs begin degrading and emitting sub-therapeutic or harmful wavelengths six months into production.
Class II Medical Device Registration is categorically different. It is a continuously enforced legal commitment across four mandatory dimensions:
Legal Accountability
Mandatory, unannounced audits of manufacturing facilities at any time — not a one-time certification event with years of unmonitored production between reviews.
Post-Market Surveillance
Strict legal obligation to trace and report any injuries or malfunctions to the relevant health authority globally. Consumer brands have zero equivalent obligation.
Clinical Substantiation
Peer-reviewed evidence that the device delivers the exact physiological results claimed — independently reviewed. Not marketing language. Documented clinical proof.
Material Biocompatibility
Every material in contact with human skin must be independently verified for non-toxicity. Consumer gadgets have no such requirement across their production lifetime.
5 Scientific Factors That Differentiate Medical Devices
Authentic clinical brands do not hide behind vague, undisclosed lab reports. Devices like Celluma are globally regulated medical tools because they adhere strictly to the validated physics and biology of Photobiomodulation:
-
Patented “Shape-Taking” Flexible Design
Physics dictates that light energy diminishes exponentially with distance (the Inverse Square Law). Rigid masks create significant energy loss at every curved area of the face — cheekbones, forehead, jawline — exactly where most visible aging occurs. Celluma's patented flexible design drapes directly against the tissue surface, eliminating all air gaps and ensuring maximum photon absorption by the mitochondria at every anatomical contour simultaneously.
-
Pulsed Delivery (PWM) — Mode-Specific
Continuous high-wattage light can cause cellular saturation and push treatment into the inhibitory zone of the Arndt-Schulz biphasic dose response. Celluma uses Pulse Width Modulation — rapidly alternating LEDs on and off at calibrated, indication-specific frequencies. This creates micro-recovery intervals that maximise ATP production while actively preventing thermal tissue damage that degrades collagen.
The Physics of Pulse Width Modulation
ON (Absorb)ON (Absorb)ON (Absorb)Pulsing delivers high-peak photonic energy followed instantly by a thermal “off” phase — preventing heat buildup while maximising bioenergetic ATP yield per session.
-
Clinical Wavelength Precision at Exact Nanometres
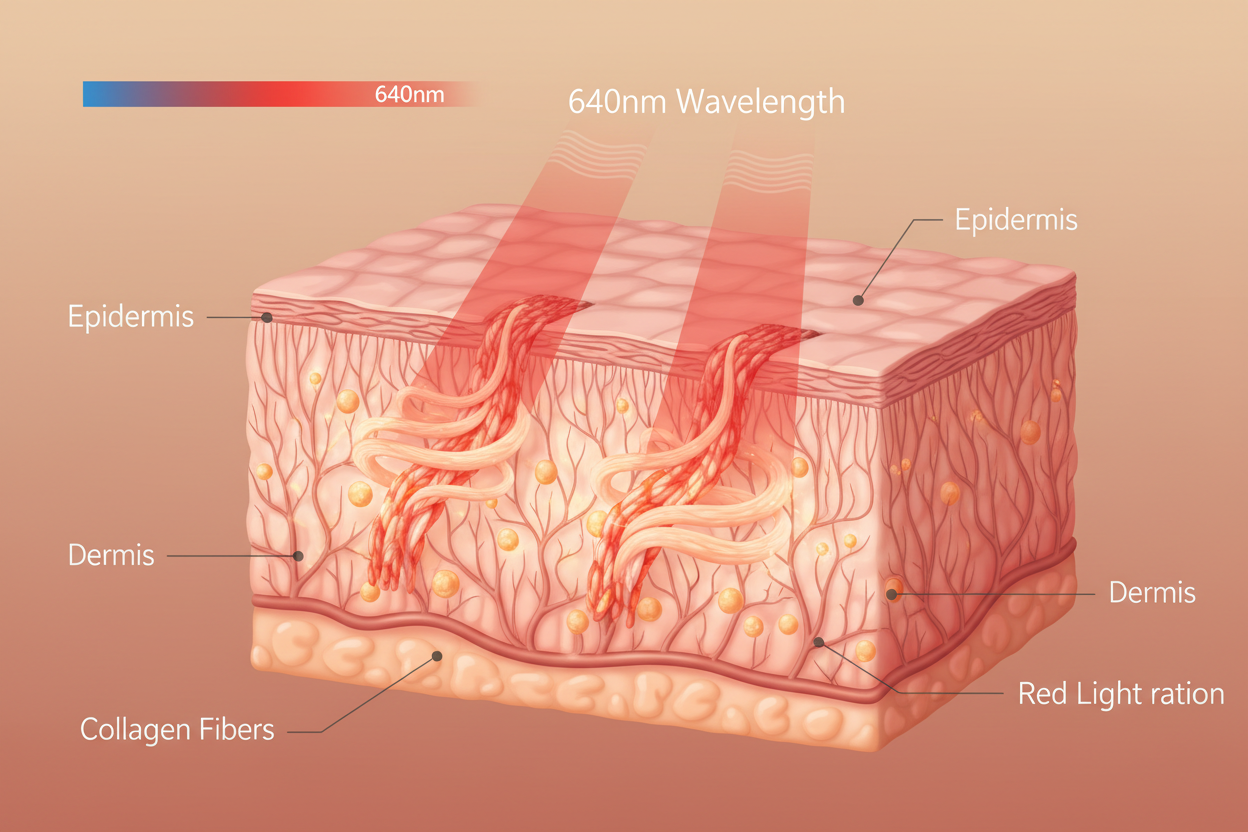
Medical devices use highly specific, independently verified wavelengths: Blue (465nm) for P. acnes bacterial destruction via Singlet Oxygen, Red (640nm) for fibroblast activation and collagen synthesis, and Near-Infrared (880nm) for deep tissue pain and inflammation. Each nanometre is confirmed by regulatory review. Consumer beauty masks emit “red light” — an unspecified range that may entirely miss the Cytochrome c Oxidase absorption peaks.
-
Uniform Energy Distribution Across the Full Panel
Cheap LED panels suffer from clustering that creates overlapping irradiance hotspots in the centre and under-dosed “dead zones” at the edges. Clinical hardware is algorithmically engineered for a perfectly uniform therapeutic dose across the entire treatment surface — every LED contributing equally, every session.
-
Verified Biocompatibility Against Human Skin
As a regulated medical device, every physical material — from the optical LEDs to the outer polymeric coating — undergoes rigorous biocompatibility testing to ensure non-toxicity against compromised or sensitised skin. Consumer gadgets have no equivalent ongoing requirement, and no obligation to investigate if sensitisation reactions are reported by users.
The Regulatory Audit: Side by Side
Across every criterion that matters for your long-term biological safety, the gap between these two standards is not a matter of degree — it is a matter of legal enforceability.
| Safety Criterion | Class II Medical Clearance FDA / CE / TGA / Health Canada |
Third-Party Tested Consumer Gadget Standard |
|---|---|---|
| Manufacturing Oversight | ✓ Ongoing unannounced factory audits | ✗ One-time snapshot only |
| Adverse Event Reporting | ✓ Legally mandated to government | ✗ Zero legal obligation |
| Clinical Efficacy Proof | ✓ Peer-reviewed human trials required | ✗ No clinical proof required |
| Wavelength Accuracy | ✓ Exact nm verified by regulator | ✗ No wavelength verification |
| Material Biocompatibility | ✓ Every skin-contact material tested | ✗ Materials unverified over time |
| UV Leakage Screening | ✓ Absence confirmed & monitored | ✗ Not assessed or tracked |
| Consumer Legal Protection | Full ongoing legal accountability | Marketing document only |
Celluma Meets Every Standard.
Four independent regulatory bodies. Continuously enforced. Verifiable at accessdata.fda.gov. Over a decade of consecutive industry recognition.
Cleared
Conformité
Goods Admin
Licensed
Regulatory Q&A
Third-party testing is a one-time snapshot with no ongoing accountability. FDA Class II clearance requires: ongoing unannounced manufacturing audits; mandatory adverse event reporting to the government; peer-reviewed clinical proof of efficacy for named indications; and rigorous material biocompatibility testing. A third-party certificate says nothing about what the device will do six months into production.
Not biologically. Third-party electrical testing confirms the device won't catch fire — it does not verify wavelength accuracy, UV leakage, material biocompatibility, or clinical efficacy. Without medical clearance, manufacturers have zero legal obligation to report injuries, degraded LED performance, or device malfunctions to any authority.
An independent FDA reviewer confirmed the device is safe and effective for specific named indications — verifiable at accessdata.fda.gov using the K-number. This is legally different from “FDA registered” (facility listing only). Cleared = independently reviewed for efficacy. Registered = address in a database.
The regulatory gap is the space between “third-party tested” (one-time marketing certification, no ongoing accountability) and “FDA Class II cleared” (continuously enforced legal standard). Brands exploit this gap by displaying lab reports that confirm electrical safety while avoiding the medical device registration process — which would require proving clinical efficacy, submitting to unannounced audits, and legally reporting adverse events.
Go to accessdata.fda.gov and search using the device's K-number (e.g. K123456). Every FDA 510(k) cleared device has a publicly verifiable record showing the device, manufacturer, cleared indications, and review date. If a brand cannot provide a K-number, the device has not been independently reviewed for efficacy.
Yes. Celluma holds FDA Class II 510(k) clearance for wrinkle reduction, acne treatment, and pain management, plus CE, TGA, and Health Canada licensing — verified independent clinical review in four jurisdictions. The K-number is publicly verifiable at accessdata.fda.gov. Celluma has won industry awards consistently for over 10 consecutive years.
Don't Settle for a Marketing Loophole.
Invest in a device that carries genuine regulatory clearance — and the legal accountability that comes with it. Available in Singapore with free island-wide delivery.
{kind=link}
Leave a comment
เว็บไซต์นี้ได้รับการคุ้มครองโดย hCaptcha และมีการนำนโยบายความเป็นส่วนตัวของ hCaptcha และข้อกำหนดในการใช้บริการมาใช้